Change Country
-or-


By John Lafferty, Life Sciences Programme Director
Read time: 9 minutes
It all started with Big Bang, since then, the universe has been expanding and randomness (variation) has been increasing, humans try to put order on our world but we constantly have to battle against the forces of nature. This, in essence, is the problem medical device manufacturers face, our devices require high degrees of order and consistency but the universe and increasing variation are against us.
The key to high-quality, consistent medical devices is controlling variation and the key to controlling variation is validation.
In this blog, I will explain how to use Process Validation to reduce variation in medical device manufacturing. If you would like to learn more about Process Validation, check out our Process Validation for Medical Devices Manufacturing – QQI Level 7 Training Course
Topics
Validation should not be regarded as something that begins at the end of the design and procurement process when everything is in place for manufacturing start up. Most progressive Medical Device companies have ceased to regard validation in this way, because this leads to protracted validation timelines, validation failures, process failures, customer complaints and disputes with suppliers. Likewise, neither can validation be left to a small number of validation specialists who are just ‘left to get on with it’.
Get it right first time by using a cross functional approach (h4)
Modern, effective validation systems utilise a cross functional team approach involving the R&D, Product Development, Production, Process Engineering, IT, QA and QC Departments working together with the Validation Department to focus on good process and equipment design and variation reduction. This approach ensures that products and processes are designed, developed and implemented in a right-first-time manner. Validation begins with process and equipment design, validation testing confirms that the approach used for design, development and implementation of the manufacturing system have indeed resulted in processes that are capable and products that consistently meet specification.
The validation approach should begin with product design and end with process and product testing.
Risk Management should wrap around the whole validation approach. Risk Analysis should feed into the product Design Inputs and each subsequent step, up to and including validation testing. Risk Analysis data should in turn be updated on the basis of testing and other information gained at each stage.
In order to realise this approach; the Validation V Model (Fig. 1) is recommended. The V Model involves documenting requirements at each of three basic stages;
Following this, the systems are built and subsequently tested by the IQ, OQ and PQ, which test the corresponding requirements as outlined in Fig. 1. below. Risk Management techniques are applied throughout this process from start to finish.
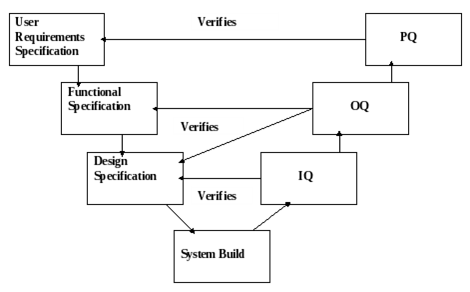
Fig. 1: Validation V Model
Fig. 1 above outlines the basic V Model, this model may be modified in order to suit the specific product, process or equipment in question. Documents may be combined or omitted as justified for any particular validation scenario.
A typical validation project might involve the steps outlined in Fig. 2. below. Product Design may well be carried out by a separate department and even at a separate site but it should not be considered to be completely divorced from the validation project, Design-for-Manufacture (DMF or DFX) is a long-established concept but sadly not always implement in practice, often due to the pressures of product launch deadlines.
However, if the principles of variation reduction are not built into product design the negative effects of this may be seen during manufacturing, often enduring for the lifetime of the product. Likewise, the Process Design and Process Development elements are not always incorporated into the validation approach again to the determent of manufacturing process consistency and product quality. Prior to commencing Product Design or Process Development, the organisation should ensure that all of the Test Methods to be used in these steps have been validated according to applicable Test Method Validation best practices. This will ensure that the product and process are designed on a sound basis. Test Method Validation at this point will also ensure that variation due to testing is minimised.
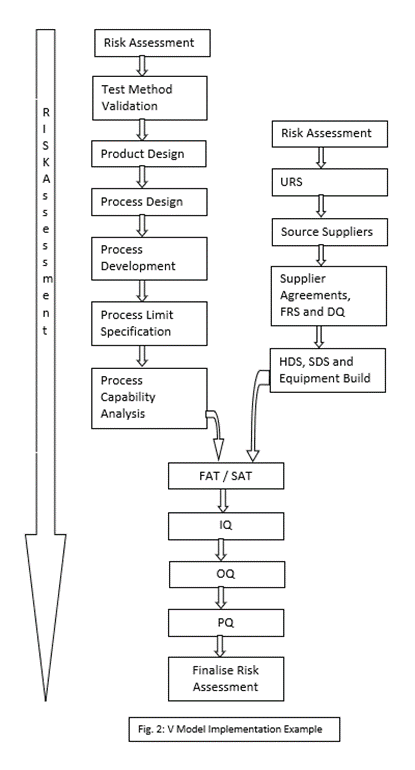
During Process Design, the Key Process Variables (KPVs) should be identified and documented. These are the input variables which have the greatest effect on the output of the process. These can subsequently be used to specify the production process.
During Process Design, the process limits should be established. These should be established by determining at what points the process begins to generate out-of-specification product. An initial Process Capability study should be carried out to ensure that the process is capable prior to transfer to production. Subsequent process validations should assess the Capability of the production process.
Establishing process limits based on +/- 3 Standard Deviations forming the Mean is not recommended. This approach does not facilitate subsequent Process Capability analysis during production nor does it prove product functionality at the process limits. Where the point of failure cannot be easily determined; testing product at +/- 4, 5 or 6 Standard Deviations may be a possible method for establishing process limits.
During the Process Development phase, a high Capability Index (Cpk) (ideally > 2) should be sought to allow for potential increases in variation in the production environment. If a Cpk of close to two is not achievable at the Process Development stage, then re-design of the process in order to reduce variation should be considered. In some cases, it may be necessary to completely re-think the manufacturing process in order to achieve the desired variation reduction. If the Cpk cannot be improved, then risk assessment should be utilised to determine what, if any, controls will be required to safeguard product performance.
Having completed Test Method Validation earlier in the validation project will pay dividends here, as the team will be able to determine how much of the variation seen is due to the process and how much is due to the test method. In some cases, improvements to test methods may be required and Gauge Repeatability and Reproducibility (Gauge R&R) studies may have to be repeated.
Find out about our Process Validation for Medical Device Manufacturing Training QQI Level 7
Any manufacturing process is hugely dependent on the quality of manufacturing equipment. Problems with manufacturing equipment can easily become protracted as a third party (the equipment supplier) is involved and very often contractual issues can arise. Proper specification of equipment, good planning of validations, choosing the right supplier and watertight agreements with suppliers are the key to preventing problems with purchased equipment.
It is recommended that the V Model approach be applied to the qualification of all purchased equipment. This is particularly important when dealing with equipment that contains software. It is recommended that the principles of GAMP 5 Ed. 2 be applied to the validation of equipment software.
Risk assessment should be used to decide whether or not to perform audits of suppliers prior to the procurement of equipment. It is recommended that equipment Design Qualification (DQ) be part of the equipment validation programme. Design Qualification involves formal checks to ensure that the equipment Functional Requirements Specification (FRS) met the requirements for the equipment (and the process) as defined in the User Requirements Specification (URS). Similarly, DQ ensures that the equipment design conforms to the concept outlined in the FRS and the requirements of the URS. For off-the-shelf equipment DQ ensures that the Functional Design Specification (FDS) meets the URS, very often the equipment Operating Manual can act as the FDS. A key focus of the DQ should be checking that the equipment is designed for cleanability, maintainability, process efficiency and above all minimisation of process variability.
The decision as to whether to perform Factory Acceptance Testing (FAT) or Site Acceptance Testing (SAT) will depend on such factors as; if the equipment is off-the-shelf or purpose built and, the expected response time from the supplier for fixing problems should they arise. The decision as whether or not to perform FAT or SAT should be documented in an agreement with the supplier. This agreement should also communicate clearly the performance levels, including process capability, that will be required of the equipment and the process, during validation and subsequent use. The supplier’s responsibilities should the desired performance not be achieved should be clearly documented.
During the FAT and SAT phases, it is essential to establish process stability and process capability. Process stability can be assessed through the use of X-bar and R Control Charts. Ideally no process should be accepted unless it is stable. Once process stability has been achieved, it is important to determine if the data to be used for Capability Analysis is Normally distributed. If the process data is expected to be Normally distributed but is not, then the process must be investigated to determine the cause of the lack of Normality and corrective must be action taken. (Note: not all process produce data that is Normally distributed) If non-normal data must be used, then statistical methods such as Distribution-of-best-fit or Transformations should be used to compensate for the lack of Normality, prior to conducting Capability Analysis.
Ideally no equipment should be accepted unless the pre-defined Process Capability levels have been achieved; however, process optimisation may not have taken place at this stage so equipment may be accepted even though the desired Capability levels have not yet been achieved. It is important to remember that once you receive equipment on site, problems that were the supplier’s problems now become your problems.
During the equipment testing phase IQ, OQ and PQ (or PPQ) a key focus should be those elements of the equipment and process that have the potential to affect the Key Process Variables, and any associated controls. During the OQ and PPQ phases process stability and process capability should again be confirmed through repeated Control Charting, Normality Testing and Process Capability Analysis. Once the reliability of the process has been demonstrated and the pre-determined Process Capability levels have been achieved during the OQ and PPQ phases, the organisation can look forward to a process that consistently produces product that meets all its specifications and quality attributes.
The key to high-quality, consistent medical devices is controlling variation and the key to controlling variation is validation. Using the variation reduction methods above will allow your organisation to minimise variation in your manufacturing processes and to produce medical devices that consistently meet specification.
Process Validation for Medical Devices Manufacturing – QQI Level 7 Training Course (6-day course)
Process Validation and Equipment Validation (3-day course)
Follow the SQT Life Sciences Page for more updates on Process Validation and Life Sciences Topics
Sign up to receive the latest industry and company news direct to your inbox.